I've been having a few problems building/checking a package in Rstudio and have been writing this so I don't suffer again. I haven't added anything to my blogger site in ages, because blogdown has taken over my technical blogging - but, since this post will involve making a package from scratch using the Rstudio witchcraft and it isn't obvious how package-creation could be done and documented within an Rmarkdown file, I'm just going to post screenshots here (and eventually move the figures over to blogdown)
This blogpost is errors included. Hopefully I'll end up with a cleaner package-setup workflow and include a TL/DR to summarise that. But I like to see where people ballsed-up and how they learned, and learning programming typically involves a lot of the former...
----
Create a new environment (under miniconda using conda-forge)
conda create --name pkg_temp -c conda-forge \
r-base=3.5.1 r-devtools=2.0.1 r-roxygen=6.1.1
conda activate pkg_temp
conda install rstudio=1.1.456
Start Rstudio
rstudio &
----
Create a new package
Within Rstudio:
File ->
New Project ->
[Create Project within a] New Directory ->
[Project Type is an] R Package -> ...
We set the package name to be "hello_world", then "new_project", then "temp_project". In each case the text-entry field went red.
It went red, because it doesn't want underscores in the package name.
So we set the package name to be "helloProject"; this adds a new subdirectory to the file system. We didn't add .git, use any existing source files or use packrat
The package is initialised with a "hello.R" file (this appears to be the default for new packages in Rstudio).

----
Get that initial package to pass `devtools::check()`
`devtools`, `usethis` and `roxygen2` are the (current) standard tools for assisting you make and document packages in R.
I loaded `devtools`, `usethis` and `roxygen2` so that they are available within the console in Rstudio, although they are implicitly available: you can build / install / test a package from the Build tab in Rstudio [in the top-right of the image below (your build tab may be elsewhere)].

We have the skeleton of a package. The package contains a single function (`hello`). Let's see what we need to do to get that package installable:

Nothing! We just clicked "Install and Restart" and the template package installed (we could similarly have used `devtools::install()` in the console).
But. If you want to release a package you have to get your package to pass R's package "check"ing workflow. At the command line you'd write `R CMD check <my_package...>`, in the console you'd use `devtools::check()` and in Rstudio you'd use `Build -> Check` or [Ctrl-Shift-E] (or similar; shortcuts are written for the linux version of Rstudio). Using `Build -> Check` on the template package we get the following:

[Choosing a license]
`Check` failed because we have yet to pick a license.
The DESCRIPTION file holds a variety of info about your package. Before we choose a license it looks like this:

I usually use the MIT license. To choose this using the console we do this:
usethis::use_mit_license(name = "My Name")

That didn't fail, but it looks like it wasn't happy (we have some warnings related to the contents of the DESCRIPTION file, but can ignore them). The DESCRIPTION now looks like this (and a couple of files called LICENSE and LICENSE.md have been added to the root directory):

We'll check the package again:

So that all seems great. We have a package containing a single function, it can be installed and it passes the checks.
----
Ensure we can update the package docs & code
The template package had an `R/hello.R` (the source code) and a `man/hello.Rd` file (the documentation for that source code). The contents look like this:
[R/hello.R]

[man/hello.Rd]

Let's change that hello.R file so that it returns a data.frame with "hello" in one column and "world" in another The code in `hello.R` now looks like:
hello <- function(){
data.frame(x = "hello", y = "world")
}
We install and check the package (both are fine). Calling `hello()` returns a data.frame but when we call `? hello` it says the function _prints_ out "Hello, world!", so the documentation is out of sync with the code.

We could modify "man/hello.Rd" to reflect the change to the `hello` function. But by using `roxygen2` we can have the content for the documentation for `hello` stored with the source code (ie in R/hello.R), and have the documentation-files that R uses (ie, man/hello.Rd) generated automatically from the source code. This simplifies development a bit.
`roxygen2` is available in our environment (see the conda stuff, above). To use it within this project, we have to tell Rstudio that we want to use `roxygen2` to generate the `*.Rd` documentation files whenever we build our package. To do this, go through `Tools -> Project Options -> Build Tools` and click "Generate documentation with roxygen" (and then click Automatically roxygenize when running "Build and reload" in addition to the existing options)
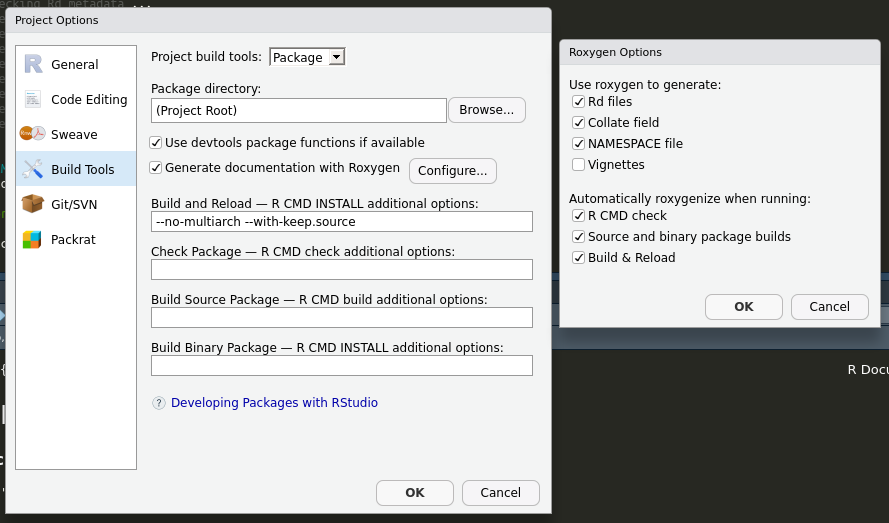
Now we can update the docs from the definition of the `hello` function within the source file `R/hello.R`.

When generating documentation with roxygen, typically you would build/install the package (with [Ctrl-Shift-B] or `Build -> Install and Restart`) or use [Ctrl-Shift-D] or `Build -> More -> Document`. In a package, `roxygen2` would then create any of the `man/*.Rd` files and update the `NAMESPACE` file (the latter indicates which packages / functions / classes are to be imported-into or exported-from the current package).
We can't do that yet. If any of the files that `roxygen2` wants to make are already in existence (and haven't been made by roxygen itself) it won't make those files. So, since `hello.Rd` and `NAMESPACE` already exist in our skeleton package, these won't be automatically made. Running the roxygen2-Documentation workflow ([ctrl-shift-D]) gives the following in our project:

Indeed, if we reload our package and call `? hello`, the help-page shows that hello should print, but it returns a data.frame.
So, we delete the existing `man/hello.Rd` and `NAMESPACE` files and rerun roxygen2-Document. It works this time:
[Ctrl-Shift-D - after deleting the existing files]

We reinstalled the package [Ctrl-Shift-B] and checked the function / help-page:

Well that's pretty weird. The function can't be called now, but the documentation can be read. The problem is that we haven't exported the function.
The original NAMESPACE file contained a rule that exported anything found in files of the form "R/*.R". But we're now using roxygen2 to both generate the documentation (which it's done) and the NAMESPACE file (which it has done, but it isn't quite how we wanted it). Aside from a comment, the NAMESPACE file is empty at the moment:
To ensure that our `hello` function is exported, you could modify NAMESPACE (adding the line `export(hello)`); but that comment is warning you not to make any changes to it. To ensure `hello` is exported, you add an `@export` roxygen annotation to the `hello`-function's docstring:

We can reinstall the package (and automatically update the NAMESPACE & documentation, since roxygen2 is now ran everytime we build the package, see above) and `hello()` should be available.
[Ctrl-Shift-B]

----
Import a function from another package (without using it)
Now that we can install, check and automatically document a package, let's break it on purpose.
We use the `roxygen2` `@importFrom` annotations to make external functions available in our new package. Suppose that, rather than using `data.frame` we wanted to use `data_frame` from the `tibble` package. There are several reasons for preferring the latter: `data_frame` doesn't automatically convert strings to factors, the returned objects have nicer default printing behaviour and it doesn't bork up your column names.
We add an `@importFrom` annotation to import `data_frame` from `tibble` and replace the call to `data.frame` with `data_frame`:
We add an `@importFrom` annotation to import `data_frame` from `tibble` and replace the call to `data.frame` with `data_frame`:

Then we document/build/reinstall (no problems), and check [Ctrl-Shift-E]. Check failed with an error "Namespace dependency not required: `tibble`".

We are trying to use a function from an external package (tibble) within our local package (helloPackage). Suppose R is loading our package into a new session, how does it know which external packages must be available, and which packages/functions to import? It looks in DESCRIPTION and NAMESPACE.
DESCRIPTION tells R which packages are required for our package to work (in addition to authorship and licensing information). During a call to `install.packages` these external packages will be installed prior to installation of our package.
NAMESPACE tells R which functions to import from those packages or, which packages to import wholesale (and explains which functions are exported from our package)

We are trying to use a function from an external package (tibble) within our local package (helloPackage). Suppose R is loading our package into a new session, how does it know which external packages must be available, and which packages/functions to import? It looks in DESCRIPTION and NAMESPACE.
DESCRIPTION tells R which packages are required for our package to work (in addition to authorship and licensing information). During a call to `install.packages` these external packages will be installed prior to installation of our package.
NAMESPACE tells R which functions to import from those packages or, which packages to import wholesale (and explains which functions are exported from our package)
So to use the `tibble` package, we should have updated DESCRIPTION and NAMESPACE. roxygen2 updates NAMESPACE automatically - as we can see in the current NAMESPACE below:


But DESCRIPTION hasn't been updated to say that our package depends upon tibble. We shouldn't try to use a function from an external package if we can't be certain that that external package is available in the environment where we are trying to use our package.

We can update DESCRIPTION by hand, or use `usethis::use_package` to ensure that R knows that `tibble` is a prerequisite for our package. We prefer the latter since it's less prone to typos. `data_frame` was first exported from version 1.0.0 of `tibble`, and from version 1.5.0 of `usethis` we can specify a minimum-version of an external package for use within a package we are developing -
After a call to:
usethis::use_package(
"tibble",
# with usethis >= 1.5.0 we can specify a minimum version:
min_version = "1.0.0"
)

We can update DESCRIPTION by hand, or use `usethis::use_package` to ensure that R knows that `tibble` is a prerequisite for our package. We prefer the latter since it's less prone to typos. `data_frame` was first exported from version 1.0.0 of `tibble`, and from version 1.5.0 of `usethis` we can specify a minimum-version of an external package for use within a package we are developing -
After a call to:
usethis::use_package(
"tibble",
# with usethis >= 1.5.0 we can specify a minimum version:
min_version = "1.0.0"
)
... our DESCRIPTION looks like this (note the format for specifying the minimum package versions):

We now document / build / install [Ctrl-Shift-B] and Check [Ctrl-Shift-E]: all pass without any issues.
----
[TODO: Pass the barebones package through styler, lintr and get it to pass all goodpractice checks (except cyclocomp)]
[TODO: Add github repo; Do the minimal to set up and pass travis integration]
----
ACK! `data_frame` is being withdrawn from `tibble` so the example running through this post is already bad practice (I could rewrite using `tibble::tibble` but that would lead to a confusion between the name of the external-package and the name of the function imported from that package)

We now document / build / install [Ctrl-Shift-B] and Check [Ctrl-Shift-E]: all pass without any issues.
----
[TODO: Pass the barebones package through styler, lintr and get it to pass all goodpractice checks (except cyclocomp)]
[TODO: Add github repo; Do the minimal to set up and pass travis integration]
----
ACK! `data_frame` is being withdrawn from `tibble` so the example running through this post is already bad practice (I could rewrite using `tibble::tibble` but that would lead to a confusion between the name of the external-package and the name of the function imported from that package)





